- R语言是一种用于统计计算和图形生成的编程语言和操作环境,主要用于数据分析、统计建模以及制图。
- 基因差异表达和GO富集分析是生物信息学中用于分析高通量测序数据的两种重要方法,通常用于理解不同条件下基因表达的变化。
一、基础定义
- R语言中的基础定义有:
- 变量:在R中,可以使用赋值运算符(<- 或者 =)来给变量赋值。变量名可以包含字母、数字、点和下划线,但不能以数字开头。
- 数据类型:R支持多种数据类型,包括数值型、字符型、逻辑型、复数等。
- 向量:向量是R中最基本的数据结构之一,是一维数组,可以存储相同类型的元素。可以用c()函数来创建一个向量。
- 矩阵:矩阵是二维的数据结构,所有元素必须是相同的类型。可以用matrix()函数创建。
- 数组:数组是多维的数据结构,与矩阵类似,但是可以有超过两维。
- 因子:因子是用于存储分类数据的向量,通常用于表示有限数量的不同组或类别。
- 列表:列表是一种复合数据结构,可以包含不同类型的对象,如向量、矩阵、列表等。
- 数据框:数据框类似于表格或电子表格,每一列可以包含不同类型的值,是R中用于存储和处理数据集的主要结构。
- 函数:R语言中的函数是一段可重复使用的代码块,用来执行特定任务。R自带了许多内置函数,并且用户也可以自定义函数。
- 包(Packages):R的扩展库被称为包,它们提供了额外的功能,比如更复杂的统计分析工具或者图形界面。你可以通过install.packages()安装新的包,并用library()加载到当前会话中。
二、基因差异表达、GO富集
- 基因差异表达是指在不同的实验条件或样本组之间比较基因表达水平,以识别那些表达显著变化的基因。
- 一旦找到了差异表达的基因列表,接下来就可以进行GO富集分析,以了解这些基因可能参与的生物学过程、分子功能和细胞组成成分。Gene Ontology(GO)是一个结构化的、受控的词汇表,描述了基因产品在生物学中的作用。
三、代码实例
0、准备
- 安装并载入所需的包:
#包的安装
install.packages("BiocManager")
library(BiocManager)
BiocManager::install("DESeq2")
install.packages("ggplot2")
library(ggplot2)
install.packages("pheatmap")
library(pheatmap)
install.packages("dplyr")
library(dplyr)
- 设置工作目录:
#0. 设置当前工作目录
setwd("xxx") # xxx为你设定的R的工作目录
#查看工作目录
getwd()
1、画图
- 从外部读取数据:
# 从外部读入数据
rpkm<-read.table("rpkm.txt",header=T,row.names=1)
head(rpkm,n=20)
head:取数据的前n行,检查数据
- 绘制箱线图:
# 用读入的RPKM数据绘制箱线图
rpkm<-read.table("rpkm.txt",header=T,row.names=1)
boxplot(rpkm)
输出:
- 美化箱线图:
log2(rpkm[,1][rpkm[,1]>0])->S0h1
log2(rpkm[,2][rpkm[,2]>0])->S0h2
boxplot(S0h1,S0h2,col=c("cyan","purple"),main="log2(RPKM)boxplot",names=c("S0h1","S0h2"))
输出: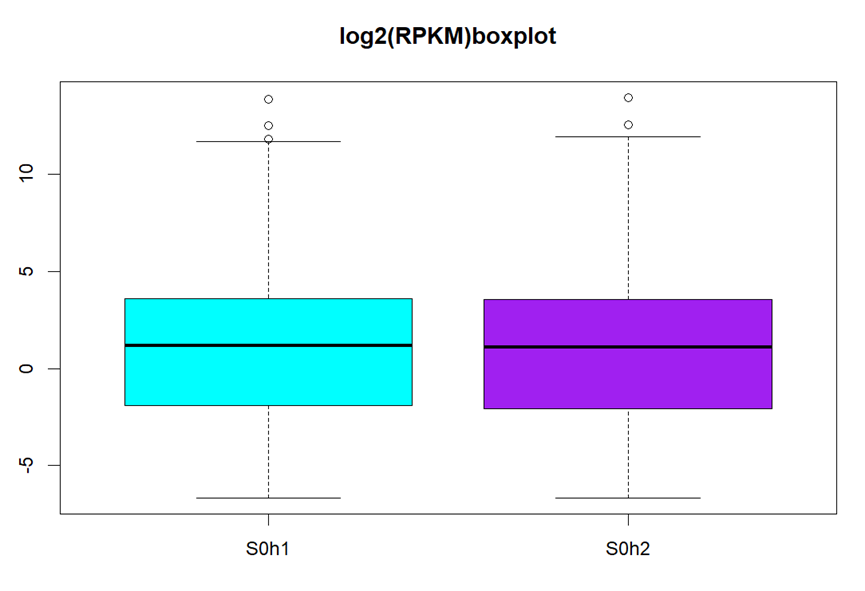
- 绘制直方图:
# 用读入的RPKM数据绘制直方图
par(mfrow=c(1,2))
hist(S0h1,breaks=100,main="log2(RPKM)histogram",col="blue")
hist(S0h2,breaks=100,main="log2(RPKM)histogram",col="red")
输出: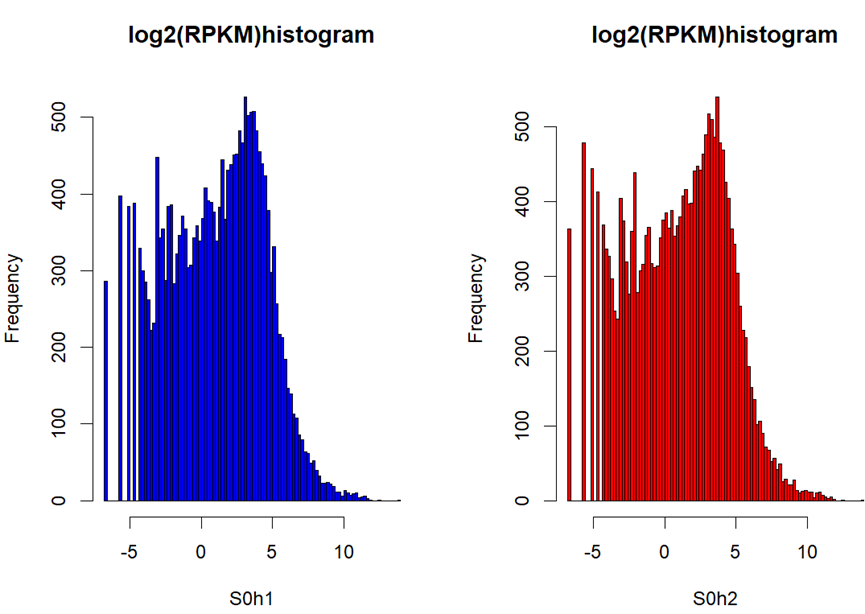
2、相关性分析
- 用R检验两重复样本的相关性:
# 用R检验两重复样本的相关性
log2(rpkm+1)->rpkm
ggplot(rpkm,aes(x=S0h1,y=S0h2))+geom_point(col="blue",cex=.6)+xlab("S0h1 log2(RPKM+1)")+ylab("S0h2 log2(RPKM+1)")->sp
model<-lm(S0h2~S0h1,rpkm)
summary(model)
sp+stat_smooth(method=lm,col="red",cex=1.1)+annotate("text",label="R^2==0.98",x=1,y=10,parse=TRUE)+theme_classic()
输出: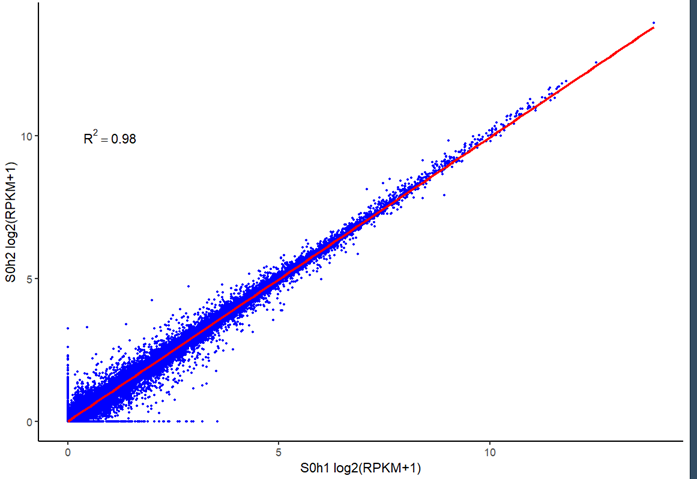
- 用R画reads染色体分布图:
# 用R画reads染色体分布图
read.table("coverage.txt",header=T,sep="\t")->coverage
head(coverage)
#将 coverage 数据框中的 chr 列转换为因子类型,并指定因子的水平为1到22号染色体,以及X和Y染色体
coverage$chr<-factor(coverage$chr,levels=c(1:22,"X","Y"))
p<-ggplot(coverage,aes(x=position,y=coverage,fill=chr))+
geom_bar(stat="identity",width=.5)+
ylab("Coverage")+
scale_fill_manual(values=rainbow(24))+
facet_grid(chr ~ .)+ #添加分面,按染色体分割图形,每个染色体一个面板
scale_y_continuous(breaks=c(0,3))+
xlab("Position")
p
# ggsave(filename="coverage_bar.png",plot=p,height=10,width=6.18) # 保存图
ggsave( )是保存图片
输出图:
3、基因差异表达分析
- 用R筛选差异表达基因并绘制火山图:
# 用R筛选差异表达基因并绘制火山图
library("DESeq2") # 载入DESeq2包
data <- read.table("genecount.txt")
dim(data)
head(data)
- 预处理数据,去掉低表达的基因:
# 预处理数据,去掉低表达的基因,只包含那些在至少一个样本中有表达的基因
# rowSums(data)计算data数据框中每一行的和
expressionData <- data[rowSums(data) >= dim(data)[2],]
dim(expressionData)
head(expressionData)
expressionData <- expressionData+1
head(expressionData)
- 创建因子dex及元数据框:
# 创建因子dex及元数据框
dex <- factor(c(rep("L2",3),rep("L3",3)))
meta <- data.frame(row.names=colnames(data),dex)
- 创建DESeqDataSet对象:
# 创建DESeqDataSet对象
dds <- DESeqDataSetFromMatrix(countData = expressionData, colData = meta, design = ~ dex)
dds <- DESeq(dds)
res <- results(dds)
head(res)
res_1 <- data.frame(res)
res_1 %>%
mutate(group =case_when(
log2FoldChange >= 2 & padj <=0.05 ~ "Up",
log2FoldChange <= -2 & padj <=0.05 ~ "Down",
TRUE ~ "Unchanged"
)) -> res_2
table(res_2$group)
- 画火山图:
# 画火山图
DEdata <- read.csv(paste('H1_vs_L1.bud.csv',sep = "."),header = T)
dt <- data.frame(DEdata, stringsAsFactors = FALSE, check.names = FALSE)
table(dt$group)
dt$log10FDR <- -log10(dt$padj)
- 获取表达差异最显著的10个基因:
# 获取表达差异最显著的10个基因;
top10up <- filter(dt,group=="Up") %>% distinct(X,.keep_all = T) %>% top_n(10,abs(log2FoldChange))
top10down <- filter(dt,group=="Down") %>% distinct(X,.keep_all = T) %>% top_n(10,abs(log2FoldChange))
top10sig <- rbind(top10up,top10down)
- 调整火山图:
# 新增一列,将Top10的差异基因标记为2,其他的标记为1;
dt$size <- case_when(!(dt$X %in% top10sig$X)~ 1,dt$X %in% top10sig$X ~ 2)
# 提取非Top10的基因表格;
df <- filter(dt,size==1)
# 指定绘图顺序,将group列转成因子型;
df$group <- factor(df$group,levels = c("Up","Down","Unchanged"),ordered = T)
# 自定义半透明颜色(红绿);
mycolor <- c("#FF99CC","#99CC00","gray80")
p <- ggplot(data=df,aes(log2FoldChange,log10FDR,color=group))+
#-----------------------------
geom_point(size=1.6)+
scale_colour_manual(name="",values=alpha(mycolor,0.7))+
geom_point(data=top10up,aes(log2FoldChange,log10FDR),color="#FF9999",size=3,alpha=0.9)+
geom_point(data=top10down,aes(log2FoldChange,log10FDR),color="#7cae00",size=3,alpha=0.9)+
labs(y="-log10FDR",title = 'H1_vs_L1')+
scale_y_continuous(expand=expansion(add = c(0, 0)))+
scale_x_continuous(limits = c(-10, 10))+
geom_hline(yintercept = c(-log10(0.05)),size = 0.7,color = "orange",lty = "dashed")+
geom_vline(xintercept = c(-2,2), size = 0.7,color = "orange",lty = "dashed")+
theme_classic()
p
输出: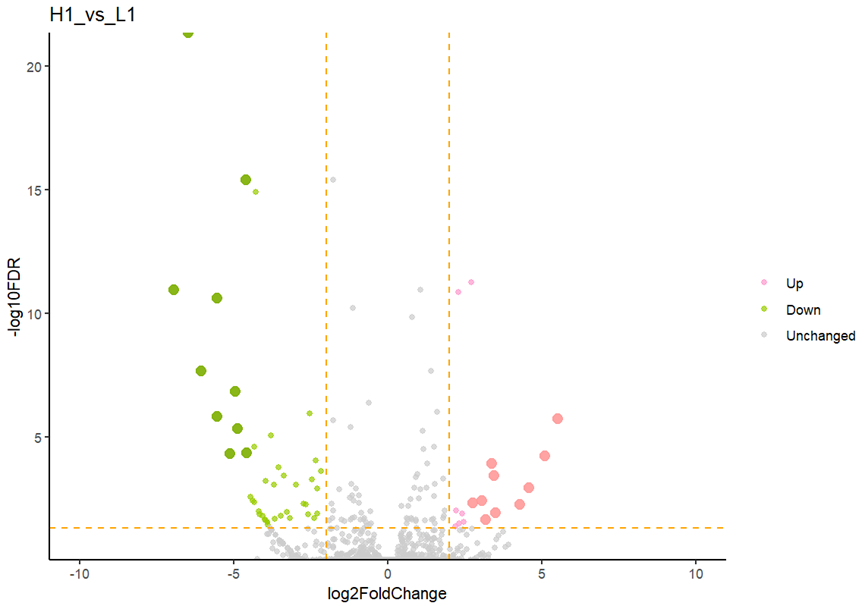
4、GO富集分析
- 用R进行聚类分析并作图:
# 用R进行聚类分析并作图
data <- read.table("heatmap.txt",header=T,sep="\t",row.names=1)
head(data)
data.m = as.matrix(data)
data.m = log2(data.m+0.01)
pheatmap(data.m,
cluster_col = TRUE, # 如果是FALSE表示只对行进行聚类
clustering_distance_rows = "correlation",
scale="row",
colorRampPalette(c("green", "black", "red"))(100),
fontsize_row=6)
输出图: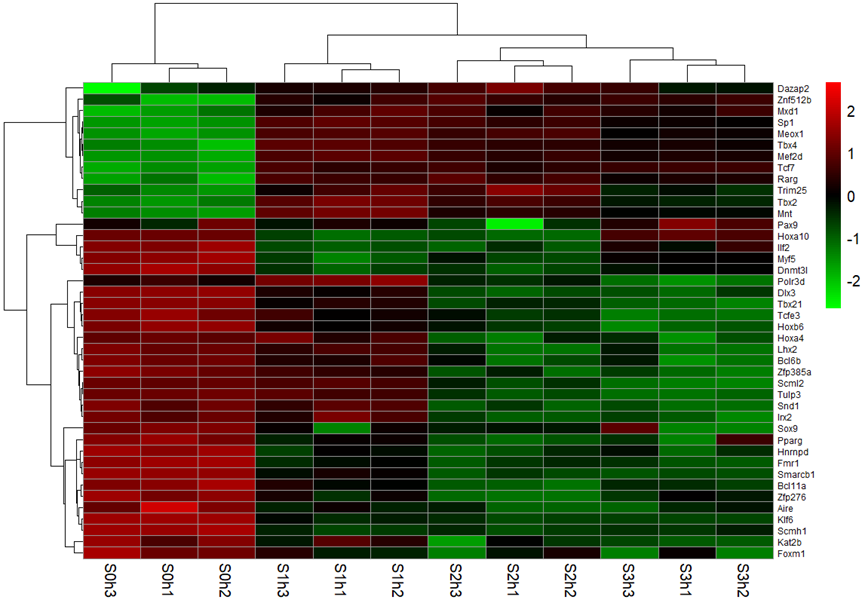
5、线性拟合
- 拟合并获取相关信息:
x <- c(151, 174, 138, 186, 128, 136, 179, 163, 152, 131)
y <- c(63, 81, 56, 91, 47, 57, 76, 72, 62, 48)
relation <- lm(y ~ x)
print(relation)
summary(relation)
输出: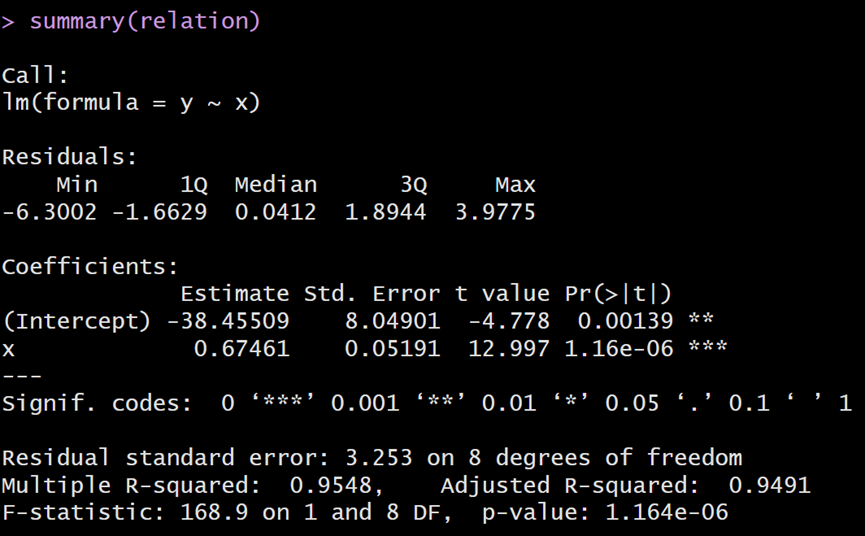
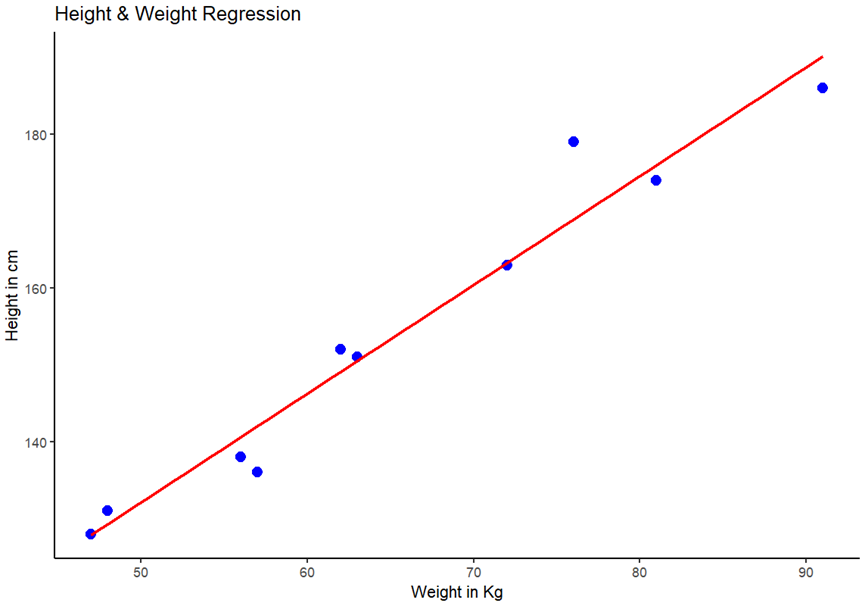
- 用ggplot画拟合图:
library(ggplot2)
data <- data.frame(weight = y, Height = x)
ggplot(data, aes(x = weight, y=Height))+
geom_point(color = "blue", size = 3)+
geom_smooth(method = "lm", color = "red", se = FALSE)+
labs(title = "Height & Weight Regression",
x = "Weight in Kg", y = "Height in cm")+
theme_classic()
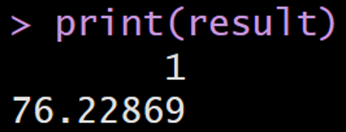
a <- data.frame(x = 170)
result <- predict(relation, a)
print(result)
输出: